



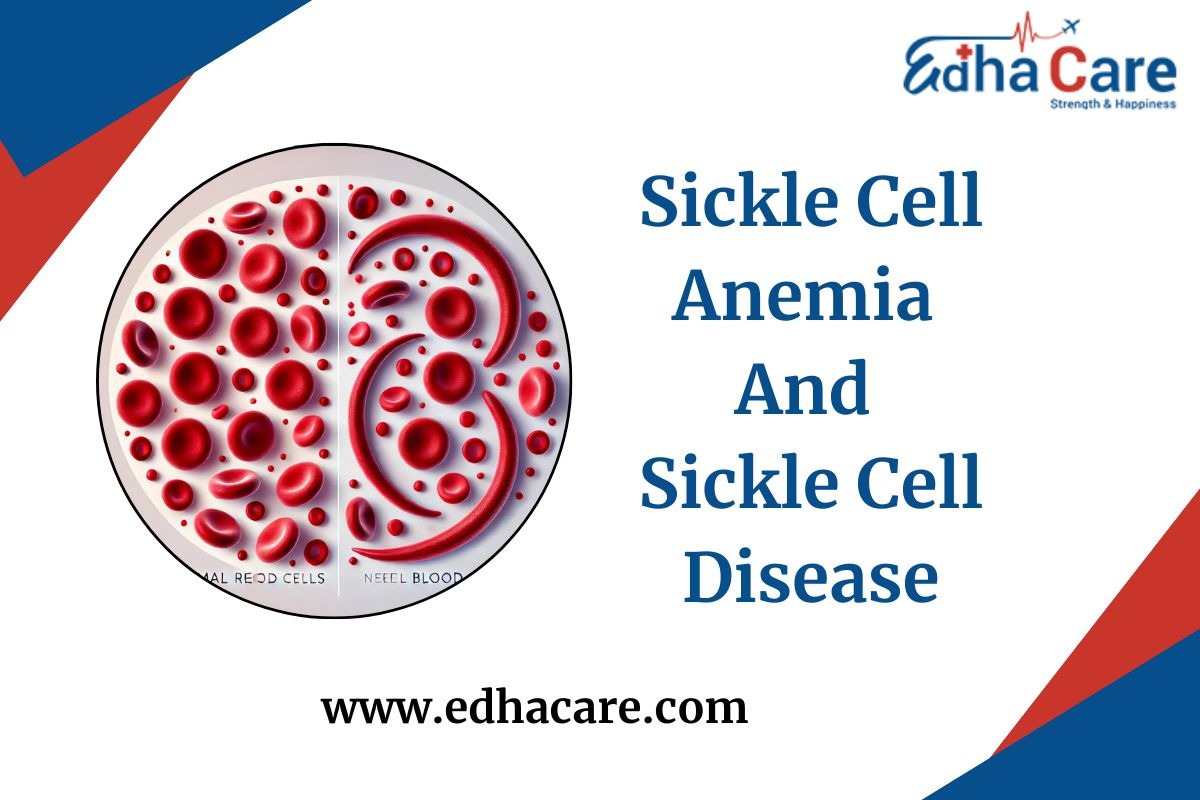
Sickle Cell Disease (SCD) is a group of inherited red blood cell disorders that affects the shape and function of red blood cells. Normally, red blood cells are round and flexible, allowing them to move easily through blood vessels. However, in Sickle Cell Anemia, the cells become rigid and shaped like a crescent or sickle, leading to blockages in blood flow and reduced oxygen delivery throughout the body.
This condition causes chronic anemia, severe pain episodes, and can lead to organ damage if not properly managed. Understanding the difference between Sickle Cell Anemia and Sickle Cell Disease is essential for early diagnosis and effective treatment.
What Is Sickle Cell Disease (SCD)?
Sickle Cell Disease refers to a group of inherited disorders in which a person has abnormal hemoglobin, known as Hemoglobin S. When this abnormal hemoglobin releases oxygen, it causes red blood cells to distort into a sickle shape. These sickled cells are sticky and tend to block blood flow, leading to various complications.
Types of Sickle Cell Disease include:
- Sickle Cell Anemia (HbSS): The most common and severe form of SCD.
- Sickle-Hemoglobin C Disease (HbSC): A milder variant with fewer complications.
- Sickle Beta-Thalassemia (HbSβ): A combination of sickle cell and thalassemia traits.
All types can cause pain crises, anemia, and other systemic issues.
Understanding Sickle Cell Anemia
Sickle Cell Anemia is the most severe form of Sickle Cell Disease. It occurs when an individual inherits two abnormal hemoglobin genes — one from each parent. The sickled red blood cells die early, causing a constant shortage of healthy cells, resulting in chronic anemia.
Common Symptoms of Sickle Cell Anemia:
- Fatigue and weakness
- Pain episodes (also called crises)
- Shortness of breath
- Delayed growth or puberty
- Frequent infections
- Swelling in hands and feet
- Vision problems
These symptoms often begin in early childhood and require lifelong management.
Complications of Sickle Cell Disease
If untreated, sickle cell disease can lead to severe health issues such as:
- Stroke
- Organ damage (especially kidneys, liver, and spleen)
- Pulmonary hypertension
- Leg ulcers
- Increased infection risk
Therefore, regular monitoring, preventive care, and early treatment are vital to improving life expectancy and quality of life.
Treatment Options for Sickle Cell Disease and Anemia
While there is currently no universal cure for SCD, medical advances have made the condition manageable.
1. Medications:
- Hydroxyurea: Helps reduce painful crises and the need for blood transfusions.
- Voxelotor and Crizanlizumab: Newer drugs that improve oxygen delivery and reduce blockages.
2. Blood Transfusions:
- Used to increase normal red blood cell count and prevent complications like stroke.
3. Bone Marrow or Stem Cell Transplant:
- The only potential cure for SCD, especially when performed with a matched donor.
4. Supportive Care:
- Pain management, hydration, and avoiding triggers such as extreme temperatures or stress.
EdhaCare partners with India’s leading hematology hospitals to provide advanced treatments like blood transfusion therapy and stem cell transplants for international patients seeking affordable and high-quality care.
Conclusion
Sickle Cell Anemia and Sickle Cell Disease are serious genetic blood disorders that require lifelong management and medical attention. Early diagnosis, continuous care, and advanced treatments can significantly improve patients’ lives.
EdhaCare connects patients worldwide to India’s best hematologists and super-specialty hospitals, offering cutting-edge therapies for blood disorders at affordable costs.
Comments